Publications
Teratology Primer, 3rd Edition
How Are New Medicines Evaluated for Developmental Toxicity?
Susan B. Laffan GlaxoSmithKline, King of Prussia, PA
Melissa S Tassinari Harwich MA
Since routine safety testing in pregnant women is generally not conducted for ethical reasons, clinicians are in the unusual situation of relying on animal data when making prescribing decisions for women who could become, or are, pregnant. Human safety data during pregnancy are rare before drug approval and are typically obtained from pregnancy registries and epidemiology studies that become available long after the medicine is approved for use. Most medicines are never tested in pregnant women. All product labeling for medicines contains a section on use in pregnancy. This section is almost always entirely composed of animal data rather than data from human clinical trials. Only women not of reproductive potential (e.g., hysterectomy or postmenopausal) or who are using effective contraception are enrolled in early clinical trials. After completion of initial clinical trials the animal studies are conducted, which would identify potential developmental or reproductive hazards. Then reproductive age women are typically included in the later, large scale clinical trials with appropriate precautions for pregnancy prevention.
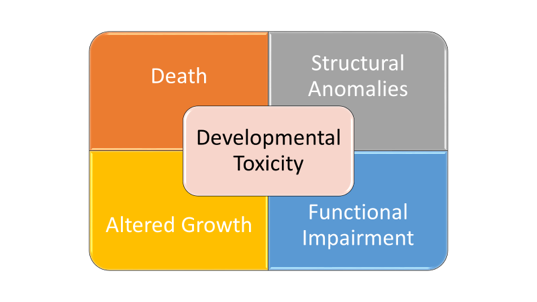
So, what kind of testing is conducted in animals? Developmental and reproductive toxicity (DART) animal studies are designed to assess safety at all stages of the life cycle, starting with reproductive capacity of adults, through offspring conception and embryofetal and postnatal development, to reproductive capacity of the offspring. Separate studies are typically conducted to cover all the life stages. Developmental toxicity is an overarching term used to describe any adverse outcome to the offspring. These studies assess four main types of developmental toxicity: death (embryo, fetal or neonatal), structural abnormalities, alterations in growth and physical maturations, and functional impairment. The category of structural abnormalities includes fetal malformations and variations. Malformations are commonly defined as fetal abnormalities judged to potentially affect survival, growth, development, functional competence, or external appearance. Thus, a teratogen is an exposure that, at a certain dose, causes malformations that can be structural and/or functional. Fetal variations are abnormalities or retardations in development, transitory alterations or permanent alterations not believed to adversely affect offspring. Functional impairment includes neurodevelopmental effects, deafness, and/or infertility. The four manifestations of developmental toxicity (death, structural anomalies, growth alterations, functional impairment) are distinct manifestations with potentially different periods and doses for susceptibility. Reproductive toxicity generally refers to effects that diminish capability to reproduce or become pregnant. Fertility in animals is assessed by treating adults prior to and during mating and in females through early embryogenesis of offspring.
Regulations
Developmental and reproductive toxicity testing of new medicines in experimental animal models is required by heath authorities. Worldwide, the International Council for Harmonization (ICH) provides guidelines for testing pharmaceuticals, which are followed by most countries within their own regulations. The studies that follow these ICH guidelines are intended to test the potential for adverse effects on males and females from adult pre conceptional exposure through exposure via the milk in newborn animals. Reproductive toxicity studies, also referred to as fertility studies, are conducted to evaluate for potential effects on reproductive capacity of adult male and females.
Figure 1. Graphic courtesy of Susan B. Laffan.
Study Designs
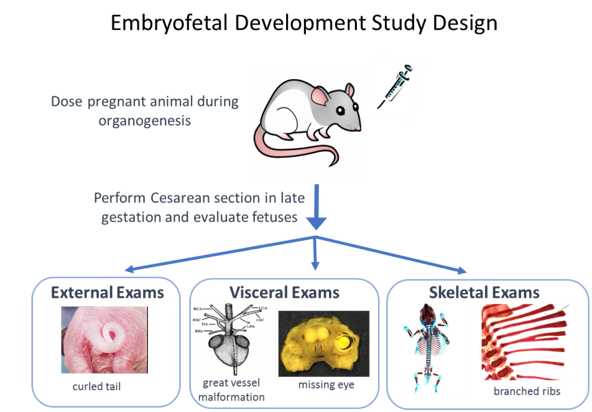
The only stage of development that is generally recommended to be tested in two species is embryofetal development. In embryofetal development studies, pregnant animals are dosed during organogenesis, from the time of implantation to closure of the hard palate, which is comparable to the human first trimester. A detailed assessment of the fetuses exposed in utero is conducted after cesarean section. Fetuses are examined externally and internally for malformations and variations of the organs (viscera) and skeleton. Growth is evaluated by body weight and in some cases by long bone length or crown rump (body) length. These studies are typically performed in one rodent and one nonrodent species. Rats with a gestational length of 21 days, and rabbits with a gestational length of 29 days are the most often used species. In the age of biologic medicines, there is increased use of primate models because the traditional rat and rabbit models are not pharmacologically relevant. To minimize animal use, the study design in primates is typically modified to an “enhanced” pre and postnatal development study in which Cesarean sections are not conducted and the offspring are delivered. The animals are examined at birth for effects of prenatal exposure and then postnatal developmental stages are assessed. All DART studies can be customized, based on properties of the medicine or theoretical concerns to include different assessments or dosing periods.
Figure 2. Graphic courtesy of Susan B. Laffan.
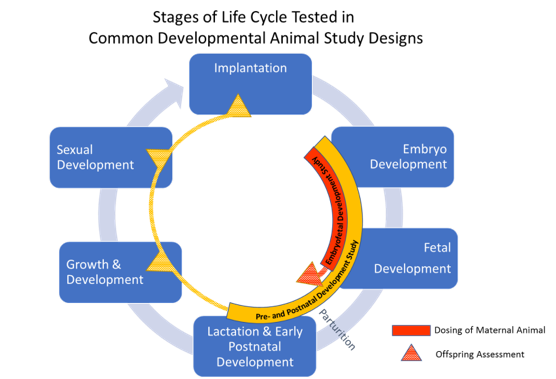
Embryo-fetal developmental toxicity studies evaluate the potential for structural malformations and developmental delays of the fetus but are not designed to assess effects on offspring function. Another ICH guideline study is used to assess function; the pre- and post-natal study includes medicine exposure during both the prenatal and early postnatal stages of development and is typically conducted in one species. In a pre and postnatal study, pregnant rats are dosed during organogenesis (pre-natal) through lactation until the day of weaning (~post-natal day 21). The treated dams are observed to evaluate the potential impact of exposure on parturition and the ability to care for the young during the lactation period. For the offspring, body weight changes are measured to evaluate growth. Offspring are observed for the onset of physical hallmarks of sexual development and primitive reflexes. Once the animals attain sexual maturity, functional reproductive ability is evaluated by mating the offspring. Once mature, the animals are put through a battery of neurobehavioral tests including tests to evaluate motor activity, reflexes, learning, and memory. These tests often involve swim mazes because rats are adept, but reluctant swimmers. The ability to learn how to get out of the swim maze is assessed to test learning skills and then later, the ability to remember the route is assessed to test memory.
Dose Selection
The dose levels tested in animal studies are carefully selected to cover a range of dose levels that provide sustainable exposures at or above expected human exposures. Animals are typically exposed to three different dose levels and outcomes are compared with a control group, which is exposed only to inactive vehicle. The medicine is tested over a range of doses up to a dose level providing exposures that are expected to cause some degree of maternal toxicity or stress. The highest dose is usually chosen as one that will produce maternal toxicity, for example, a small decrease in pregnancy weight gain or reduced food consumption. The low dose is chosen to provide an exposure in the animal close to the anticipated human exposure level. The lowest dose level of the medicine that produces developmental toxicity is called the LOAEL (lowest observed adverse effect level). The highest dose level that does not cause developmental toxicity is the NOAEL (no observed adverse effect level). These levels can be compared to the anticipated human exposure level.
Data Interpretation
Completion of these developmental and reproductive (DART) studies is only half the task; appropriate interpretation of these data is key and should be done by scientists trained in the concepts and principles of developmental toxicity. When interpreting DART studies, it is extremely important to know if developmental toxicity is related to pharmacological activity and if it occurred in the absence or presence of maternal toxicity. Maternal illness or stress in a dam could affect her offspring resulting in secondary effects (e.g., reduced survival, developmental delays in growth and maturation). The presence of developmental toxicity in the absence of any maternal toxicity is of more concern in assessing risk as it implies the hazard has occurred outside of any maternal influence and is a direct result of the medicine. Teratogenicity is a concerning findings as it is typically not affected by maternal toxicity. It is also important to know the background incidences of malformations in animal models to determine the effects of a medicine. Data are reviewed to determine if there is a dose response relationship, meaning that the incidence and severity of adverse outcome increases with increasing dose. Assessing patterns is important in assessing risk. A study that shows no effect of low and high doses, but shows an effect at a middle dose, is less convincing than a study that relates increasing effect to increasing dose. Understanding the pharmacologic action of the medicine and the mechanisms for toxicity are also important.
Figure 3. Graphic courtesy of Susan B. Laffan.
Risk Determination
The data from DART animal studies form the basis for assessing risk during human pregnancy. Data from animal models provide a scientifically valid and ethical assessment of assessment of potential hazards for humans, to be put in context with the risk or probability of the effect related to the pharmaceutical use. The ability of a medicine to cause developmental toxicity is usually related to the systemic exposure, (i.e., the concentration in blood and tissues) and timing of exposure during critical stages of embryofetal development. For some medicines, brief pulses of high exposure during a specific developmental stage will produce toxicity, whereas other medicines may require a lower, but longer duration of exposure to produce toxicity. Medicines that cause developmental toxicity in one species may cause developmental toxicity in another species, although the specific manifestations may be different. If both species have effects, it raises the level of concern for risk to human pregnancy. For instance, if the anticipated human plasma exposure level is 100 times lower than the NOAEL of the animal study (based on plasma exposure in the animals), adverse effects on human development are considered unlikely. Some researchers believe that a 100 times “safety margin” is unnecessary, and consider 25 or 50 times safety margins as adequate in most situations. However, the severity of the developmental toxicity observed must be considered. Study outcomes including fetal death or malformations (spina bifida, gastroschisis, limb defects, etc.) are somewhat rare and raise the level of concern. It is much more common for studies to indicate effects on overall development at maternally toxic dose levels, such as lower fetal body weights, delayed skeletal ossification, and an increase in incidence of early embryofetal loss in a litter (i.e., increased postimplantation loss). In instances where there are no abnormal effects on the offspring, even when the medicine is given at maternally toxic dose levels, the evidence suggests that abnormal effects on human development are unlikely. Any testing scheme, however, cannot categorically define a medicinal product as safe or unsafe because this expectation ignores the importance of the exposure level in determining toxicity.
Finally, the prescriber, along with the patient must weigh the risks against the benefits of the medication. A medicine that is a member of a class known to be teratogenic at relatively low exposure levels in animal experiments would carry a warning against use in pregnancy, but in a pregnant woman with a serious disease, it may be important to use the medicine despite the possible risk. In these circumstances, animal data remains an important part of the risk evaluation to characterize the margin of safety for humans with respect to dose level and possibly the critical window of sensitivity with respect to timing of pregnancy. In other words, human embryos and fetuses are not uniquely sensitive provided that the medicine has been tested at sufficiently high doses in experimental animals. There are medicines that produce abnormal development at the high doses used in experimental animal studies but not at the exposure levels encountered by humans. Experimental animal testing is designed to be conservative for use of medicines in pregnancy. Again, the entire benefit versus risk must be considered for each patient.
Conclusion
While human data on developmental toxicity is increasingly included in pharmaceutical labeling, most the medicines approved for use still have no human data in the pregnancy section of their labeling. Consequently, we must rely on animal data in the product labeling to understand potential risk for both newly approved and existing medicines. Developmental toxicity studies in animals provide informative but complex data for assessing potential risks of medicine use in human pregnancy. The animal data along with individual factors, including genetic background and the risks of the underlying disease being treated are weighed to make rational decisions about the use of any medicine during pregnancy.
Suggested Reading
Beyer BK, Chernoff N, Danielsson BR,Davis-Bruno K, Harrouk W,Hood RD, Janer G, Liminga UW, Kim JH, Rocca M, Rogers J, and Scialli AR. (2011) ILSI/HESI maternal toxicity workshop summary: Maternal toxicity and its impact on study design and data interpretation. Birth Defects Research Part B – Developmental and Reproductive Toxicology. 92(1):36–51.
ICH Guideline for Industry S5(R3):. Detection Of Toxicity To Reproduction For Human Pharmacueticals. Step 2 Draft 2017. http://www.ich.org/
Carney EW and Kimmel CA, (2007) Interpretation of Skeletal Variations for Human Risk Assessment: Delayed Ossification and Wavy Ribs. Birth Defects Research (Part B)- Developmental and Reproductive Toxicology 80:473–496.
Hood RD, Developmental and Reproductive Toxicology 2nd edition. Florida; CRC Press, Taylor & Francis Group, 2006.
Lo WY and Friedman J M: Teratogenicity of Recently Introduced Medications in Human Pregnancy. Obstet. Gynecol. 2002: 100:465–473.
Rogers JM. Developmental Toxicology. In Casarett and Doull’s Toxicology 8th edition, Klaassen CD (ed.) New York; McGraw Hill, pp 481–524, 2013.
Shepard TH. Catalog of Teratogenic Medicines 13th edition. Baltimore; The Johns Hopkins University Press, 2011.
Schardein JL. Chemically induced Birth Defects 3d edition. New York; Marcel Dekker, Inc 2000.
Scialli AR, Buelke-Sam JL, Chambers CD, et al.: Communicating Risks During Pregnancy: A Workshop on the Use of Data from Animal Developmental Toxicity Studies in Pregnancy Labels for Medicines. Birth Defects Research Part A 2004: 70: