Publications
Teratology Primer, 3rd Edition
How Has Epigenetics Influenced Understanding of Teratogenesis?
Robert M. Greene, Ph.D.
Department of Surgical & Hospital Dentistry, Division of Craniofacial Development & Anomalies, University of Louisville, Louisville, KY
M. Michele Pisano, Ph.D.
Department of Surgical & Hospital Dentistry, Division of Craniofacial Development & Anomalies, Department of Pharmacology & Toxicology, University of Louisville, Louisville, KY
Developmental defects are complex and heterogeneous in nature due to contributions from, and interactions between, genetic and environmental risk factors. Despite significant progress in the identification of genetic and environmental risk factors, the majority of congenital anomalies still have no known cause. For many specific anomalies, emphasis has been placed on the search for single-nucleotide polymorphisms (SNPs), where there is a DNA sequence variation, deletion or addition of a single nucleotide, within a gene. It has become apparent, however, that the genome is also organized in an entirely different plane, 'above' the level of DNA sequence — at the epigenetic level. Epigenetics represents a field of study that examines modifications to the genome that alter the activity of certain genes in the absence of changes in the nucleotide sequence. Some of these epigenetic modifications are heritable. Current biological thought now recognizes that epigenetics is a fundamental contributing process in embryogenesis, and that the environment (nutrition, stress, chemical exposures including cigarette smoke, and heavy metals etc.) can have a profound effect on shaping the epigenome. Epigenetic alterations that could mediate many of these environmental insults during embryonic or fetal development include 1) methylation, where methyl groups are added to the DNA molecule, 2) altered expression of microRNAs (miRNAs), small RNA molecules that directly regulate gene expression without themselves being translated into proteins, 3) modification (phosphorylation, acetylation, methylation etc) of histone proteins, and 4) remodeling of chromatin via histone modification or restructuring of nucleosomes (DNA segments wrapped around histone protein cores) , among others. Environmentally-induced epigenetic alterations have the contrasting properties of being both plastic (modifiable) and mitotically stable. Thus, an environmentally-induced epigenetic alteration can be replicated during cell division and faithfully transmitted through the cell’s lineage with potentially profound negative consequences for development of tissue and embryonic structures in which such cells are found. Thus, the term epigenetics has come to be defined as the study of individual mitotically stable structural modifications to genes that alter their activity without changing the DNA sequence. Epigenomics refers to the global study of epigenetic changes across the entire genome.
Conventional screening techniques such as mutational analysis or genome-wide association studies (GWAS) do not identify epigenetic alterations or epigenetically altered genes. Such analyses focus almost solely on protein-encoding genes, which constitute a mere 1.2% of the human genome, leaving the remaining 98.8% of the genome largely unexplored. The knowledge that environmental perturbations trigger cellular reprogramming, with downstream effects on cellular function and phenotypes, has led to the development of epigenome-wide association studies (EWAS). Such studies hold the promise of identification of epigenetic factors that contribute to the etiology of congenital malformations and human disease conditions. Since the developmental periods of preimplantation and organogenesis are particular susceptibility to epigenetic influences, epigenetic alterations may represent key mechanisms in the misregulation of gene expression during embryogenesis and present the intriguing notion that such alterations may be potential loci, undetected by mutational or GWAS analyses, for congenital defects. Indeed, some congenital syndromes (e.g. Prader-Willi, Beckwith-Wiedemann and Angelman syndromes) can be caused by factors that utilize epigenetic mechanisms to produce abnormal expression of genetic loci critical for normal embryogenesis. Moreover, it is well documented that mutations of genes involved in epigenetic mechanisms, such as DNA methyltransferases and histone deacetylases, have been linked to several human syndromes (Rett syndrome, Rubinstein–Taybi syndrome).
Three independent epigenetic mechanisms that regulate gene expression serve to illustrate:
DNA methylation:
Methylated DNA is reflective of regions where a methyl group (−CH3) has been added to cytosine nucleotides. Methylation usually involves cytosines followed by guanines, often in clusters called CpG islands. Such methylated regions normally are found in areas that are transcriptionally silent, whereas unmethylated regions generally correspond to regions of DNA active in transcription. Advances in high-throughput DNA analysis now enable examination of epigenetic variants, such as changes in DNA methylation patterns across the genome. As such, EWAS provide an opportunity to detect aberrant methylation patterns at a population level, allowing association with disease phenotypes and congenital defects. While direct DNA promoter methylation generally has been thought to result in gene silencing, genome-wide methylation profiling has provided the understanding that methylation changes also can alter distal regulatory elements, particularly enhancers. Enzymes responsible for DNA methylation are known as DNA methyltransferases (DNMTs). One type, DNMT1, maintains methylation at preexisting sites. In mammalian semiconservative DNA replication, DNMT1 recognizes the methylation of the parental strand of DNA and matches it to the new daughter strand. Thus, modifications of DNA methylation patterns induced by exposure to environmental conditions during embryogenesis can become heritable. Since genomic programming is critical for normal embryogenesis, such developmental reprogramming can potentially result in abnormal embryonic development. Prenatal exposure to cigarette smoke, alcohol, or heavy metals are examples of environmental exposures eliciting epigenetic changes during development.
MicroRNAs:
While DNA methylation is the best studied epigenetic process, the ability of small non-protein-coding RNAs, specifically microRNAs (miRNAs), to regulate gene expression has come under increasing scrutiny as an epigenetic phenomenon that appears to play a significant role in embryonic development. Genes that code for such RNAs account for nearly 90% of the human genome. The discovery of these vast conserved genomic stretches encoding for non-protein-coding regulatory RNAs has revealed a previously unrecognized layer of genomic information of potentially fundamental importance to embryonic development, creating a far reaching paradigm shift regarding how our "genetic code" functions. MicroRNAs, representing only one type of non-protein-coding RNA, are 20 to 22 base pairs long and downregulate gene expression via complementary binding to the 3-prime untranslated region of target RNAs. Using orofacial clefting as an example, compelling hints that DNA sequence variations in human genes encoding miRNAs, or miRNA processing enzymes, were able to discriminate between humans with orofacial clefts and those without a cleft phenotype. This provides support for the premise that dysregulation of miRNAs might underlie the etiology of nonsyndromic cleft palate. Additional support comes from the association of a SNP in the human gene encoding miR-140 with isolated cleft palate, and the demonstration that SNPs in the miRNA binding sites of MSX1, a candidate gene for susceptibility to nonsyndromic orofacial clefts (NSOC), are also associated with NSOC susceptibility. Collectively, these data provide 1) convincing evidence that specific miRNAs play a role in orofacial development and 2) support for the intriguing notion that miRNAs, might be potential gene loci for congenital orofacial defects and may serve as biomarkers to diagnose NSOCs.
Histone Modifications:
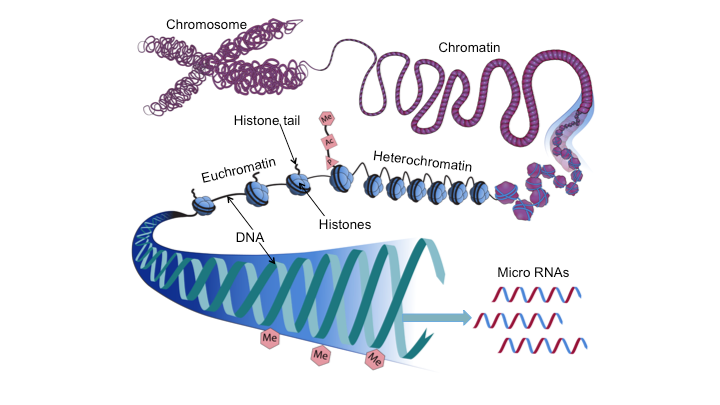
DNA is usually wrapped around histone proteins forming chromatin (Fig.1). Remodeling of chromatin via postranslational modifications of histones and alterations in chromatin structure represent two additional epigenetic means by which gene expression is regulated. In general, chromosomal regions are either tightly packed (heterochromatin) and genetically inactive, or are loosely configured (euchromatin) which represents single-copy, transcriptionally active DNA (Fig.1). Post-translational modification of histones include methylation, acetylation, phosphorylation, and sumoylation. Histone acetylation, for example, is thought to be associated with relaxation of chromatin, facilitating access to transcription factors, and enhancing gene transcription. This may provide mechanistic insight into how aberrant acetylation or deacetylation of histones may contribute to the formation of Fragile X or Rubinstein-Taybi syndromes. Histone methylation, by contrast, is considered to be associated with tightly packed chromatin, impeding access of transcription factors to DNA and, as such inhibiting transcription. Such histone modifications are thought to fine-tune the regulation of specific genes.
Figure 1. Diagrammatic representation of selected epigenetic mechanisms that may alter the epigenome and affect embryonic development and effect teratogenesis. DNA associates with histone proteins to form chromatin. Epigenetic processes including methylation (Me), acetylation (Ac), or phosphorylation (P), to name a few, can alter the epigenetic profile of histone tails, as well as the extent to which DNA is wrapped around histones (blue spheres). Chromosomal regions can be tightly packed (heterochromatin) and genetically inactive, or loosely configured (euchromatin) and transcriptionally active.
Abundant evidence exists, confirmed in many populations as well as in animal models, linking in utero environmental exposures to increased susceptibilities and adult onset conditions including hypertension, coronary heart disease, obesity, metabolic syndrome, and type II diabetes. This phenomenon is now referred to as “developmental origins of health and disease” (DOHD) or the Barker hypothesis, after David J. P. Barker, who proposed such a linkage in 1990. The original hypothesis posited that adverse in utero conditions/exposures, with birth size serving as a response marker of the intrauterine environment, program the fetus to have metabolic characteristics that can lead to future disease. Such “programming” is currently thought to take the form of epigenomic alterations and illustrates the developmental plasticity of the embryo/fetus.
Conclusion
Epigenetic regulation is fundamental to tissue differentiation and normal embryonic development. The field of epigenetics has provided new insights into the molecular underpinnings of the fetal origins of adult diseases and teratogenesis. Large amounts of genomewide data are being generated for the interdependent fields of gene expression, genotyping, and epigenomic alteration. Each is being explored for associations with diseases and, increasingly, congenital anomalies. A challenge facing teratologists is to determine the means to integrate these datasets to reveal how variations in any one impact the others such that normal embryonic development is perturbed.
Suggested Reading
Berdasco M and Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet 132:359–383 (2013).
Flanagan JM. Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol Biol. 1238:51-63 (2015).
Gluckman PD, Hanson MA and Low FM.The role of developmental plasticity and epigenetics in human health. Birth Defects Research (Part C) 93:12–18 (2011).
Friedman JM. Using genomics for birth defects epidemiology: Can epigenetics cut the GxE gordian knot? Birth Defects Res. (Part A), 91:986-989 (2011).
Ivorra C, Fraga MF, Bayón GF, et al., DNA methylation patterns in newborns exposed to tobacco in utero. J Transl Med. 13:25 (2015).
Krauss RS, Hong M. Gene-environment interactions and the etiology of birth defects. Curr Top Dev Biol. 116:569-80 (2016).
Lappalainen T, Greally JM.Associating cellular epigenetic models with human phenotypes. Nat Rev Genet. 18:441-451 (2017).
Li J, Zou J, Li Q, Chen L, et al., Assessment of differentially expressed plasma microRNAs in nonsyndromic cleft palate and nonsyndromic cleft lip with cleft palate. Oncotarget. 7:86266-86279 (2016).
Rodger EJ and Chatterjee A. The epigenomic basis of common diseases. Clinical Epigenetics 9:5 (2017).
Skinner MK. Role of epigenetics in developmental biology and transgenerational inheritance. Birth Defects Research (Part C) 93:51–55 (2011).
Strahl BD and Allis CD. The language of covalent histone modifications. Nature, 403, 41–45 (2000).
Turner BM. Histone acetylation as an epigenetic determinant of longterm transcriptional competence. Cell Mol Life Sci 54:21–31 (1998).
Zou J, Li J, Ji C, Li Q, Guo X. Expression profile of plasma microRNAs in nonsyndromic cleft lip and their clinical significance as biomarkers. Biomedicine & Pharmacotherapy 82:459–466 (2016).